|
Description of the algorithm
|
Zeolite Frameworks
Zeolites
are a class of crystalline aluminosilicates
both naturally occurring and synthesized in the laboratory. They
have found many uses in industrial processes, including catalytic cracking
of alkanes, air separation, and ion exchange.

There are many different types of zeolites,
but the common feature is a structural similarity.
Typically, a zeolite has a framework composed of tetrahedrally-coordinated
atoms (T-atoms) joined by oxygens. The T-atom is usually silicon,
but substitutions with metals (Al, P,...) are common and often desired.
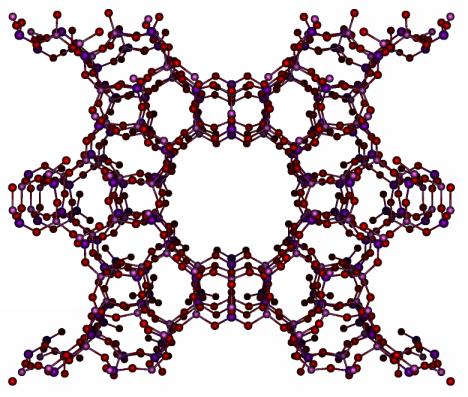
The main feature of zeolite frameworks is the presence of pores, as can
be seen in the example. These pores are 4-12 Angstrom in size and,
due to the crystalline nature of the material, can be hundreds
of Angstroms in length. Some frameworks have one-dimensional pores,
some have two or three-dimensional pores, which can join at cage-like intersections.
This makes zeolites very interesting as catalytic agents, since the bulk
of the crystal is accessible to chemical species that fit into the pores.
Metal substitutions into the framework, Al for Si for example, will create
areas of excess negative charge, where cations or non-framework species
can bind.
Structure Solution
The derivation of an atomic-scale model of
the framework crystal structure of a newly-synthesized zeolite is a non-trivial
task. The difficulty stems primarily from the polycrystalline
nature of most zeolite samples, with crystallite sizes typically below
5 um. Zeolites continue to be synthesized at a furious pace. Crucial
to the development of the field of zeolite science is the ability to determine
the structure of newly-synthesized materials: Structure is sought after
not only to understand the performance of newly synthesized catalysts but
also to propose rational syntheses of homologous materials with tailored
performance. Roughly 125 framework structures have been reported,
yet several dozen distinct synthetic zeolites remain unsolved in the patent
literature. The techniques of diversity synthesis have recently been
introduced to the field, and this may soon lead to a
tremendous explosion in the number of new, unsolved synthetic zeolites.
ZEFSA II
ZEFSA II is a direct, real-space method
for zeolite structure solution from powder diffraction data [1,2,3].
Unit cell size, density, and symmetry
are assumed to have already been determined from the powder diffraction
data. The method then locates the positions
of the T-atoms in the unit cell. The oxygens can subsequently be located
by a Rietveld refinement. The approach employs an empirical potential of
mean force for the tetrahedral atoms in a zeolite framework. ZEFSA
II is an improved method that makes use of powerful new ideas from
Monte Carlo algorithm design [3]. ZEFSA II
is able to solve all current publicly-known zeolites, starting from a high
quality dataset. ZEFSA II should be able to determine the
structure of any new single-phase zeolite for which a good powder diffraction
pattern is available.
The key step in ZEFSA II is to define
a cost function that is a function of the atomic positions within the crystalline
unit cell and that is minimized by the structure corresponding to the experimental
material. A combination of simulated annealing and biased Monte Carlo is
able to minimize the cost function and so to solve the structure
in most cases. In the most difficult cases, the superior method of parallel
tempering rather than simulated annealing is necessary. Roughly 10%
of the known zeolites require the method of parallel tempering. The
parallel tempering option is no more difficult to use than is the simulated
annealing.
ZEFSA II is freely available from
this web site under the GNU
public license. We ask that any publications making using of
the method cite the original literature [1,2,3].
1) M. W. Deem and J.
M. Newsam, Nature 342, 260-262 (1989).
2) M. W. Deem and J.
M. Newsam, J. Am. Chem. Soc. 114, 7189-7198 (1992).
3) M. Falcioni and
M. W. Deem. J. Chem. Phys. 110, 1754-1766 (1999) (download PDF file 383Kb).
ZEFSA II Home
- Description
- Data Req
- Download
- Instructions
- References
- Copying
- Structures
- Feedback